Открытый в 1954 году, синдром Дубина-Джонсона является наследственным, рецидивирующим, доброкачественным расстройством метаболизма билирубина. Это редкое аутосомно-рецессивное состояние характеризуется конъюгированной гипербилирубинемией с нормальными уровнями трансаминаз печени, уникальным образцом экскреции метаболитов копропорфиринов и осаждением пигмента, который придает печени характерный черный цвет.
Основной причиной развития синдрома Дубина-Джонсона является мутация в гене, кодирующем апикальный канальцевый мембранный белок, который несет ответственность за выведение билирубина и других соединений. Этот белок первоначально был назван органическим канальцевым транспортером анионов (также, он известен под названием белок множественной лекарственной устойчивости 2 (MRP2)). Этот белок является членом надсемейства транспортеров АВС.
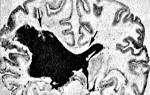
 и нормальная, здоровая печень (справа)")
Печень при синдроме Дубина-Джонсона (слева) и нормальная, здоровая печень (справа)
Начало клинического развития синдрома Дубина-Джонсона чаще всего отмечается в раннем и в позднем подростковом возрасте. Однако, в некоторых случаях, эти проявления можно зафиксировать и в неонатальном возрасте. Наследственную гипербилирубинемию можно разделить на связанную и свободную формы:
- Конъюгированная гипербилирубинемия: синдром Дубина-Джонсона и синдром Ротора
- Неконъюгированная гипербилирубинемия: синдромы Жильбера и Криглера-Наджара
Связанные и неконъюгированные гипербилирубинемии также могут быть разделены на непосредственно и косвенно реакционные формы. Непосредственно реакуионные — это те, при которых билирубин будет быстро реагировать с диазотированной сульфаниловой кислотой, а косвенно реакционные — это те, при которых билирубин также будет реагировать с этой кислотой, но очень медленно (в отсутствие акселераторов, например этанола). Эти типы гипербилирубинемий имеют относительно доброкачественное течение.
Синдром Дубина-Джонсона. Эпидемиология
Общая распространенность синдрома Дубина-Джонсона является крайне низкой. Синдром Дубина-Джонсона может развиваться у лиц всех национальностей, этнических групп и рас. Самая высокая распространенность заболевания (1 случай на 1300) была зафиксированна у иранских евреев.
Синдром Дубина-Джонсона развивается у обоих полов, но некоторые исследователи сообщают об увеличении заболеваемости у мужчин.
Синдром Дубина-Джонсона редко обнаруживается до полового созревания, хотя некоторые случаи были зарегистрированы у новорожденных. Он наиболее часто диагностируется в позднем подростковом возрасте и в начале взрослой жизни.
Синдром Дубина-Джонсона. Причины
Синдром Дубина-Джонсона является аутосомно-рецессивным расстойством, которое вызывается мутацией в гене, ответственном за выработку человеческого канальцеивого полиспецифичного органического транспортера анионов. Этот белок имеет важное значение в транспортировке некоторых органических анионов через мембрану гепатоцитов. Сам ген, кодирующий этот белок, расположен в локусе 10q24.
- Читайте также:
Читайте также: Мкб 10 синдром слабости синусового узла
Конъюгированная гипербилирубинемия, которая присутствует при синдроме Дубина-Джонсона, приводит к развитию дефектов в транспортировке билирубина-глюкуронида через мембрану, отделяющую гепатоциты от желчных канальцев. Пигмент, который не будет секретироваться из гепатоцитов, будет храниться в лизосомах и это, тем самым, приведет к окрашиванию печени в черный цвет.
Отличительной чертой синдрома Дубина-Джонсона, является специфический механизм, который полностью еще не раскрыт. У людей с этим синдромом, обычно меняется соотношение между побочными продуктами биосинтеза гема. Уровни копропорфирина I будут выше уровней копропорфирина III. А у здоровых лиц, отношение копропорфирина III с копропорфирином I составляет примерно 3-4:1.
MRP2
MRP2 играет важную роль в детоксикации многих лекарств путем транспортировки широкого спектра соединений, особенно конъюгатов глутатиона, глюкуроната и сульфатов, которые все вместе известны как продукты биотрансформации II фазы. В отличие от других членов MRP семейства, MRP2 экспрессируется только на апикальном мембранном домене поляризованных клеток.
Энергия, получаемая из АТФ, имеет решающее значение для секреторной функции MRP2. Мутации в АТФ-связывающей области MRP2 представляют собой значительную часть всех генетических дефектов, которые встречаются у лиц с синдромом Дубина-Джонсона.
Мутации
Миссенс мутации приводят к потере двух аминокислот из второго АТФ-связывающего домена MRP2. При этой мутации, будет высвобождаться мутантный белок, который не сможет транспортировать соединения из эндоплазматического ретикулума к канальцевой мембране гепатоцитов.
Синдром Дубина-Джонсона. Симптомы и проявления
Пациенты, с синдромом Дубина-Джонсона, как правило, развивают желтуху уже в подростковом возрасте. И хотя большинство пациентов имеют бессимптомное течение этого синдрома, некоторые больные жалуются на неспецифические боли в правом верхнем квадранте, это может вызвать тревогу у врачей.
- Читайте также:
Субклинические случаи могут стать очевидными после приема препаратов, которые, как известно, ухудшают транспортировку органических анионов до или во время беременности.
Отчеты многих исследований подтверждают, что пациенты с синдромом Дубина-Джонсона, могут иметь гемолитические болезни (например, наследственный сфероцитоз, талассемии), которые могут еще сильнее ухудшить желтуху.
Желтуха является наиболее ярким клиническим признаком синдрома Дубина-Джонсона. Помимо этого, результаты физикального обследования, как правило, нормальные, за исключением возможного наличия гепатоспленомегалии.
Синдром Дубина-Джонсона. Диагностика
- Синдром Дубина-Джонсона следует рассматривать у всех лиц с повышенными уровнями билирубина и с нормальными результатами тестов функционирования печени. Диагноз может быть подтвержден увеличением соотношения копропорфирина I к копропорфирину III.
- Результаты других лабораторных тестов, в том числе проверка уровней ферментов печени, сывороточного альбумина, ретикулоцитов, как правило, находятся в пределах референтных значений.
- Протромбиновое время, как правило, в пределах нормы, но оно может быть увеличено у иранских евреев с сопутствующим дефицитом фактора VII.
- Снижение протромбина наблюдается у 60% пациентов с синдромом Дубина-Джонсона.
Копропорфирины
Копропорфирины являются побочными продуктами биосинтеза гема. Обычно, копропорфирин I, преимущественно, выделяется с желчью, в то время как копропорфирин III выводится с мочой. У больных синдромом Дубина-Джонсона, выведение изомеров копропорфирина в моче будет иметь довольно уникальный рисунок, эта особенность может быть использована в качестве специфического признака, но только тогда, когда врожденные эритропоэтические порфирии и отравление мышьяком будут исключены. 80% мочевого копропорфирина составляет I тип, по сравнению с 25% у здоровых лиц.
Также интересно то, что в течение первых 2-х дней жизни, здоровые новорожденные имеют отношение копропорфиринов аналогичное тому, которое наблюдается у пациентов с синдромом Дубина-Джонсона, однако, через 10 дней жизни эти уровни выходят на нормальные значения.
Визуализация
Сцинтиграфия
Пациенты с синдромом Дубина-Джонсона, как правило, имеют уникальные гепатобилиарные проявления. В частности, сразу после внутривенного введения радиофармпрепарата (красителя), структура печени будет оставаться интенсивной и однородной в течение до 120 минут.
Интенсивная визуализация желчного пузыря может задерживаться до 90 минут после инъекции красителя, в то время как у здоровых людей, эта задержка не превышает 30 минут.
Это сочетание интенсивности и длительности визуализации печени и желчного пузыря является уникальным знаком для синдрома Дубина-Джонсона, в сравнении с другими аномалиями печени. Однако, у некоторых пациентов эти проявления могут быть ошибочно приняты за болезни желчного пузыря (это иногда становилось причиной проведения ненужной холецистэктомии).
Гистология
Макроскопически, пигмент способен окрашивать печень в темный или почти в черный цвет.
Микроскопически, у лиц с этим синдромом происходит накопление крупнозернистого пигмента, который наиболее выражен в центродолевых зонах. Рубцы, гепатоцеллюлярный некроз или искажение зональной архитектуры не наблюдается у пациентов с этим синдромом. Количество пигмента может варьироваться. Некоторые заболевания (например, вирусный гепатит) могут вызвать исчезновение этого пигмента.
Синдром Дубина-Джонсона. Лечение
Синдром Дубина-Джонсона является доброкачественным расстройством и оно не требует каких-либо специфических терапий, хотя пациенты должны быть предупреждены, что беременность, использование некоторых препаратов и наличие сопутствующих заболеваний могут усугубить желтуху.
В прошлом, у пациентов, которые принимали фенобарбитал, снижался уровень билирубина. Но эта методика уже больше не рекомендуется к применению. Сегодня, некоторые врачи применяют рифампицин и урсодезоксихолевую кислоту, которые оказывают благотворное воздействие на хронические холестатические заболевания. Эти препараты могут привести, в частности, к индукции экспрессии MRP2 в печени и почках. Но их следует применять с осторожностью. У некоторых пациентов может произойти резкое увеличение концентрации желчных кислот в сыворотке крови. Эти побочные эффекты могут возникнуть в результате повышенной экспрессии MRP3 на базолатеральной мембране гепатоцитов.
Синдром Дубина-Джонсона. Осложнения
Осложнения синдрома Дубина-Джонсона включают желтуху и гепатомегалию. Снижение активности протромбина может привести к понижению уровней фактора VII, которое встречается у 60% пациентов.
Синдром Дубина-Джонсона. Прогноз
Синдром Дубина-Джонсона является доброкачественным расстройством, а средняя продолжительность жизни пациентов, с этим синдромом, также является нормальной.
- Читайте также: